Biologie Moléculaire
La biologie moléculaire, c'est un peu de la cuisine mais en conditions nanométriques. Voici un aperçu de quelques principes et techniques de base
- PCR (Polymerase Chain Reaction)
- Coder en base 4
- Western Blot
- Méthode ELISA (Enzyme-Linked Immuno Assay)
- Séquençage de l'ADN
- Electrophorèse
- Isolation de l'ADN de plantes deshydratées
- Extraction d'ADN de cellules végétales
- Dosage des chlorophylles
- Tampons d'extraction
PCR (Polymerase Chain Reaction)
La PCR ou réaction de polymérisation en chaine, est un des éléments favoris de la boîte à outils des biologistes au laboratoire.
A la fois très simple et d'une grande élégance, elle permet d'amplifier des séquences d'acides nucléiques variés.
Avant de commencer
Comme toutes les sciences, la biologie à sa propre langue, donc pour se comprendre il nous faut défricher un peu le vocabulaire.
Voici quelques clefs :
-les acides nucléiques sont les supports de l'information génétique. Ils sont constitués de 4 briques universelles différentes nommées nucléotides (A, C, G et T/U). Agencées en séquence à brin unique (ARN) ou à double brin (ADN), ces briques représentent une information qui peut être recopiée (c'est le principe de la PCR) ou bien décodée par une machinerie spécifique à base d'enzymes. Ces séquences parfois extrêmement longues sont fragiles et souvent enroulées de manière poussée sur elles-mêmes et autour de bobines protéiques.
-la dénaturation est un processus qui vise à linéariser au mieux les séquences d'acides nucléiques pour les rendre accessibles aux enzymes. Dans le cas des ADN double brins, ces derniers s'écartent partiellement en deux brins uniques reliés par quelques points (comme les anneaux d'une chaines), donnant accès à chaque brin individuellement.
-l’appariement est le principe de complémentarité des 4 différents nucléotides dans les séquences double brin d'ADN. Il se fait d'un brin à l'autre entre nucléotides complémentaires qui se font face. C'est assez simple car il n'y a en fait qu'un appariement possible pour chaque nucléotide : le A avec le T, et le C avec le G ! Donc par exemple pour une courte séquence monobrin GATTACA, le brin complémentaire est CTAATGT.
-les enzymes sont des machines à base de protéines hyper-spécialisées, extrêmement efficientes et très diversifiées. Elles sont généralement spécifiques à un petit nombre de molécules (nommé substrat) sur lesquelles elles se fixent pour réaliser des opérations simples mais précises, avec un faible taux d'erreur. Par exemples elles peuvent tronçonner une molécule, en fusionner deux bout à bout, gérer du stockage, libérer de l'énergie, ou dans le cas de la TAQpolymérase qui nous intéresse ici, réaliser la copie d'une séquence.
-la TAQpolymérase est l'enzyme utilisée pour réaliser la PCR. Elle se lie spécifiquement aux séquences d'acides nucléiques monobrin et cherche a recréer le brin manquant.
Les étapes
En quoi consiste la PCR : une enzyme nommée polymérase va venir faire des très nombreuses copies d'une séquence modèle d'acides nucléiques.
On réalise plusieurs étapes à des températures différentes dans un appareil appelé thermocycleur. Le mélange réactionnel (Taq polymérase, Mg2+ et parfois MgCl2, amorces, les 4 dNTP en excès (A, T, C, G) et l’extrait d’ADN cible, tampon réactionnel ) est placé dans des microtubes et est soumis, plusieurs dizaines de fois, à des cycles successifs. Le thermocycleur permet la programmation de chaque étape des cycles. La réaction PCR dure quelques heures (2 à 3 heures pour une PCR de 30 cycles).
Le nombre de cycles est typiquement inclus entre 25-35 cycles d'amplification. Plus on effectue de cycles, plus on augmente le risque d'avoir des contaminants au sein de la réaction. Il est donc conseillé d'effectuer le nombre minimum de cycles nécessaire afin de synthétiser la quantité de produit désirée.
Le protocole diffère en fonction du type de PCR à effectuer :
PCR classique :
1 : Étapes du cycle PCR
Dénaturation thermique de l'ADN à 95°C : permet de rompre les liaisons hydrogène afin d'obtenir des matrices simple brin.
Hybridation des amorces à 50°C-65°C : Les deux amorces contenues en large excès s'hybrident lorsqu'elles rencontrent les séquences complémentaires. La température permet aux liaisons de se reformer. Ainsi, plus la température d'hybridation est levée, plus l'hybridation est sélective et spécifique.
Élongation à 72°C. La Taq polymérase catalyse la réplication à partir des ADN monocaténaires amorcés de façon sélective (sélectivité qui découle du choix des amorces).
Exemple de conditions de cycles pour une Taq polymérase.
| Étape |
Température |
Temps (min) |
Cycles |
| Dénaturation initiale |
95°C |
2 |
1 |
| Dénaturation |
95°C |
0,5 à 1 |
25 à 35 |
| Hybridation |
42°C à 65°C |
0,5 à 1 |
- |
| Amplification |
72°C |
1 |
- |
| Amplification finale |
72°C |
5 |
1 |
| Trempage |
4°C |
indéfini |
1 |
2 : Électrophorèse sur gel d'agarose : Ce principe repose sur l'attraction des acides nucléiques chargés négativement sous l'effet d'un champ électrique. Les acides nucléiques migrent plus ou moins loin à travers le gel en fonction de la masse des molécules (plus elles migrent loin plus elles sont petites car le maillage du gel se rétrécit plus on se rapproche du pôle +)
3 : Révélation : L'ADN est révélé par une coloration au Bromure d'éthidium ou un intercalant visible sous lampe UV (l'intercalant se lie aux acides nucléiques et émet une fluorescence)
PCR en temps réel :
À l'inverse du PCR classique, le PCR en temps réel permet une quantification plus précise grâce à une mesure de l'amplification tout a long de la réaction. On effectue la détection et la quantification de l'amplification à chaque fin de cycle grâce à un signal fluorescent, un intercalant des acides nucléiques (donc la valeur de la fluorescence est corrélée à la quantité de produit amplifié). Cette détection élimine le besoin d'électrophorèse sur gel, ce qui permet d'économiser du temps et de réduire le potentiel de contamination.
Vu que la formation de produit peut être détectée dans la phase exponentielle du PCR lorsque la réaction est la plus efficace, la quantification du matériel de départ est plus sensible et précise.
Comparaison PCR Classique et PCR temps réel
| PCR classique |
PCR en temps réel |
| Visualisation du produit amplifié par gel d'agarose après sa production |
Visualisation du produit amplifié en temps réel (pendant l'amplification) grâce à un instrument de détection |
| Ne mesure pas précisément l'ADN ou ARN de départ |
Permet de mesurer le nombre de copies d'ADN ou d'ARN de départ (d'où le nom de PCR "quantitatif") |
| Moins coûteux, ne nécessite pas d'instrument spécial |
Plus coûteux, nécessite des instruments spéciaux |
| Principe basique de biologie moléculaire |
Demande des compétences plus techniques |
Facteurs pouvant affecter le succès de la technique de PCR :
- Quantité d'enzyme utilisée
- Erreur de pipettage.
- Concentration de magnésium (Mg2+)
Purification d'ADN :
Actuellement, l'analyse par multiplex et PCR en temps réel marque l'importance de la qualité de la purification de l'ADN.
Étapes de base de la purification ADN : Lyser, lier, rincer, éluer.
| Lyse : perturbation des cellules ou tissus |
- Traitement enzymatique - Perturbation mécanique - Traitement à un détergent |
| Lyse : dénaturation ou inactivation des protéines |
- Détergents ioniques, chaleur, agents réducteurs, urée et guanidine - Protéases (ex : protéasine K) |
| Lyse : inactiver les nucléases endogènes |
- Agents perturbateurs (ex : EDTA) - Protéases (ex : protéasine K) |
| Lier et Rincer : séparer l'ADN * |
- Éliminer les autres acides nucléiques (ARN) - Éliminer les protéines, par extraction organique, grâce au sel, ou par liaison a un support solide ( matrice de silice, colonnes d'échanges anioniques, particules magnétiques). |
* Liaison et rinçage : techniques de séparation de l'ADN d'autres matériaux cellulaires.
Extraction organique : Lorsque du phénol ou une mixture de phénol/chloroforme est mélangée à un lysat cellulaire, deux phases se forment : une phase aqueuse et une phase organique. Les parts polaires de l'ADN et divisent dans la phase aqueuse, et les protéines dénaturées et d'autres débris cellulaires se divisent dans la phase organique
Extraction grâce au sel : Les sels déshydratent les protéines, ce qui les rendent moins hydrophiles et donc dénaturées. Les protéines dénaturées perdent de leur solubilité et précipitent, ces protéines dénaturées et les débris cellulaires sont extraits par centrifugation. Les sels sont typiquement utilisés dans des protocoles incluant du chlorure de sodium, potassium, acétate ou acétate d'ammonium.
Par liaison à un support solide : La plupart des techniques de purification d'ADN sont basées sur la purification des lysats bruts par une liaison sélective à un support solide. Ces supports incluent matrice de silice, colonnes d'échanges anioniques, particules magnétiques. Généralement ces méthodes sont plus rapides et plus pratiques par rapport à d'autres techniques car elles ne requièrent pas de solvants organiques et peuvent être miniaturisées et automatisées.
Sources : Molecular biolgy, Lab Guide : Promega
Fiche Technique de la PCR
Définition
La PCR (Polymerase Chain Reaction) est une méthode permettant de copier et d'amplifier une séquence spécifique d'ADN de manière exponentielle.
Principe
La PCR repose sur l'utilisation de cycles répétitifs de dénaturation, d'hybridation et d'extension pour amplifier une séquence d'ADN cible.
Composants
-
ADN matrice: Échantillon d'ADN contenant la séquence cible à amplifier.
-
Amorces: Courtes séquences d'ADN complémentaires aux extrémités de la séquence cible.
-
dNTPs: Nucléotides libres (dATP, dTTP, dCTP, dGTP) nécessaires pour l'élongation.
-
ADN polymérase thermostable: Enzyme qui synthétise le nouvel ADN, comme la Taq polymérase.
-
Tampon: Maintient les conditions de pH et d'ions pour l'activité optimale de l'enzyme.
-
Ions magnésium (Mg²⁺): Co-facteur essentiel pour l'activité de l'ADN polymérase.
Étapes de la PCR
-
Dénaturation (94-98°C): Séparation des brins d'ADN double hélice en simples brins.
-
Hybridation (50-65°C): Liaison des amorces aux séquences complémentaires sur les brins d'ADN simple brin.
-
Élongation (72°C): Synthèse de nouveaux brins d'ADN par l'ADN polymérase en utilisant les dNTPs.
Cycle Typique de la PCR
Un cycle de PCR comprend les trois étapes ci-dessus et est répété généralement 25-35 fois.
Applications
-
Diagnostic médical: Détection de pathogènes, mutations génétiques.
-
Recherche en génétique: Clonage de gènes, étude de mutations.
-
Médecine légale: Identification d'individus à partir de traces biologiques.
-
Études évolutives: Comparaison de séquences génétiques entre espèces.
Schéma de la PCR
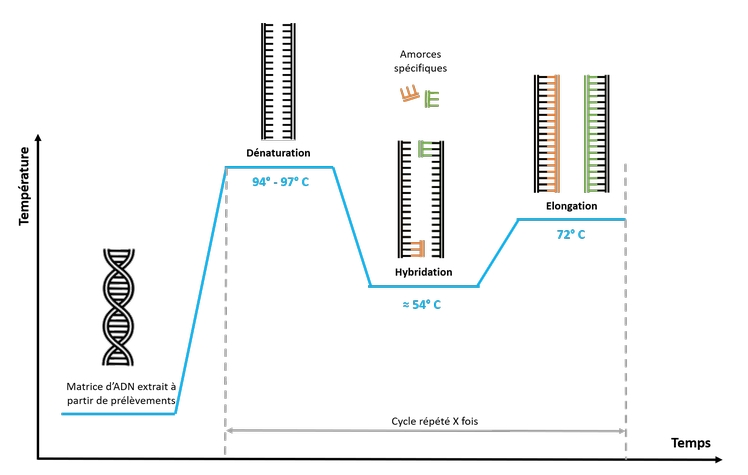
Tableau Résumé des Étapes de la PCR
|
Étape |
Température (°C) |
Durée |
Description |
|---|---|---|---|
|
Dénaturation |
94-98 |
30 secondes |
Séparation des brins d'ADN double brin |
|
Hybridation |
50-65 |
30 secondes |
Liaison des amorces aux séquences cibles |
|
Élongation |
72 |
1 minute/kb |
Synthèse de l'ADN par l'ADN polymérase |
Optimisation de la PCR
-
Température des amorces: Ajustement pour une hybridation spécifique.
-
Concentration des composants: Optimisation des dNTPs, amorces, et Mg²⁺.
-
Nombre de cycles: Adaptation en fonction de la quantité d'ADN initiale et de l'application.
Variantes de la PCR
-
qPCR (PCR en temps réel): Quantification de l'ADN en temps réel.
-
RT-PCR: Amplification de l'ADN complémentaire (cDNA) à partir de l'ARN.
-
PCR multiplex: Amplification simultanée de plusieurs cibles dans un même échantillon.
Liens et Articles Scientifiques
-
Article de base sur la PCR: Mullis et Faloona, 1987
-
PCR en temps réel (qPCR): Heid et al., 1996
-
Applications médicales de la PCR: Kalayjian et al., 1993
-
Optimisation des conditions de PCR: Dieffenbach et Dveksler, 1995
-
Utilisation de la PCR en médecine légale: Gill et al., 1985
Coder en base 4
La biologie est assez rarement binaire, elle oscille entre les notions de discret et de continu de la physique.
Toutefois, en ce qui concerne la conservation et le transfert de l'information, le support génétique universel à la vie sur terre (les séquences d'acides nucléiques ADN et ARN) est interprété par la machinerie cellulaire en code à base 3 (les triplets de nucléotides). Chacune des 3 positions successives du triplet peut présenter l'un des 4 nucléotides (A, C, G ou T/U), donc 4^3 soit 64 possibilités de triplets.
Ces triplets lus correspondent à des mots spécifiques du langage de la machinerie protéique qui doit interpréter (quel acide aminé employer dans la réalisation de la chaine, quelle action réaliser...).
Western Blot
Introduction
Le Western Blot également connu sous le nom d'immunoblot, est une technique de biologie moléculaire largement utilisée pour détecter et analyser spécifiquement des protéines dans un échantillon biologique. Cette méthode combine électrophorèse sur gel pour séparer les protéines en fonction de leur poids moléculaire et l’immunomarquage pour détecter les protéines d’intérêt a l'aide d'anticorps spécifiques.
Principes de fonctionnement

- Électrophorèse sur gel:
Les protéines sont séparées en fonction de leur taille sur un gel de polyacrylamide sous l'influence d'un champ électrique. Les protéines plus petites migrent plus rapidement a travers le gel que les protéines plus grandes. - Transfert sur membrane:
Après l'électrophorèse, les protéines sont transférées du gel sur une membrane de nitrocellulose ou PVDF. Ce transfert permet de fixer les protéines sur la membrane pour une détection ultérieure. - Détection des protéines:
La membrane est incubée avec des anticorps primaires spécifiques de la protéine ciblée, suivis d'anticorps secondaires conjugués à des enzymes ou des fluorophores. L’interaction entre les anticorps et les protéines cibles permet leur détection par des méthodes de visualisation telles que la luminescence ou la coloration.
Applications
- Analyse de l'expression protéique:
Le Western Blot permet de quantifier les niveaux d'expression des protéines dans différents types de cellules ou de tissus, offrant ainsi des informations sur la régulation génique et les voies de signalisation cellulaires. - Identification des isoformes:
Cette technique peut être utilisée pour distinguer les différentes isoformes ou formes post-traductionnelles d'une protéine en fonction de leur taille et de leur réactivité avec des anticorps spécifiques. - Détection et modification post-traductionnelles:
Le Western Blot peut être utilisé pour détecter des modifications telles que la phosphorylation, la glycosylation des protéines, fournissant ainsi des informations sur leur régulation et leur fonction. - Diagnostique médicale:
Cette méthode est largement utilisée en diagnostic médical pour détecter la présence de protéines spécifiques associées à des maladies, telles que les marqueurs tumoraux dans le cancer.
Méthodologie

I. Préparation des échantillons

Extraction des protéines:
- Lyser les cellules ou les tissus dans un tampon de lyse approprié contenant des inhibiteurs de protéases.
- Centrifuger à haute vitesse pour éliminer les débris cellulaires.
Quantification des protéines:
- Utiliser une méthode de dosage des protéines telles que le dosage colorimétrique de Bradford ou le dosage BCA.
Préparation des échantillons:
- Charger une quantité égale de protéines (généralement 20-40 µg) par échantillon dans un tampon de chargement contenant un agent réducteur et un agent tensioactif.
II. Électrophorèse sur gel

Préparation des gels :
- Préparer des gels de polyacrylamide de la concentration appropriée (généralement 8-15%) selon le poids moléculaire attendu des protéines cibles.
- Réaliser une électrophorèse en conditions réductrices.
Chargement des échantillons :
- Charger les échantillons dans les puits du gel en utilisant des pipettes à gel appropriées.
Électrophorèse :
- Faire fonctionner les gels à une tension constante jusqu'à ce que les protéines soient séparées selon leur poids moléculaire.
III. Transfert sur membrane

Équilibration du gel:
- Trempage dans une solution d'équilibre contenant du SDS pour dénaturer les protéines.
Transfert électrophorétique:
- Transférer les protéines du gel sur une membrane de nitrocellulose ou PVDF en utilisant une électrophorèse semi-sèche ou humide.
Validation du transfert:
- Vérifier l'efficacité du transfert en marquant la membrane avec une coloration protéique comme le Ponceau S.
IV. Blocage et incubation avec les anticorps

Blocage non spécifique:
- Incuber la membrane dans une solution de blocage (par exemple, lait écrémé ou BSA) pour saturer les sites non spécifiques.
Incubation avec les anticorps primaires:
- Incuber la membrane avec l'anticorps primaire spécifique de la protéine cible à une concentration appropriée et dans des conditions optimales.
Lavage:
- Laver la membrane pour éliminer les anticorps non liés.
V. Détection des protéines

Incubation avec les anticorps secondaires:
- Incuber la membrane avec un anticorps secondaire conjugué à une enzyme ou un fluorophore.
Développement:
- Révéler les protéines cibles en utilisant un substrat chromogène ou luminescent pour l'enzyme conjuguée.
Imagerie:
- Capturer les images des protéines révélées à l'aide d'un système d'imagerie approprié comme une caméra CCD.
VI. Analyse et interprétation des résultats

Quantification:
- Analyser l'intensité des bandes protéiques pour quantifier l'expression relative des protéines cibles.
Contrôle de charge:
- Utiliser des marques de poids moléculaire pour estimer les tailles des protéines cibles.
Analyse des données:
- Utiliser un logiciel d'imagerie pour analyser les données et comparer les niveaux d'expression des protéines entre les échantillons.
Remarque: Assurez-vous de suivre les protocoles de sécurité appropriés lors de la manipulation des produits chimiques et des échantillons biologiques
Conclusion
Le Western Blot est une technique puissante permettant la détection et la quantification des protéines spécifiques dans un échantillon biologique. Une bonne planification expérimentale, une manipulation précise des échantillons et une interprétation appropriée des résultats sont essentielles pour obtenir des données fiables et significatives.
Méthode ELISA (Enzyme-Linked Immuno Assay)
- Présentation -
La méthode ELISA consiste à doser visuellement une molécule présente dans une solution : un anticorps. Pour faire simple : on choisit une molécule capable de se lier avec celle qu'on veut doser : l'antigène, qu'on dépose dans un bain / un puits. On ajoute ensuite dans ce même bain la solution contenant l'anticorps. Les antigènes et les anticorps vont alors se lier. On prend soin d'ailleurs de rincer le support entre chaque manipulation pour enlever l'excédent d'anticorps appliqué. Généralement, on intègre un deuxième anticorps capable de produire de la couleur ou de la lumière (grâce à une enzyme). Enfin, l'ajout d'un substrat permet de provoquer la réaction lumineuse avec l'enzyme. La concentration en couleur perçue (à l’œil, au microscope, au colorimètre ou au spectrophotomètre...) permet alors directement de :
- conclure sur la présence ou non de l'anticorps dans la solution testée
- obtenir une estimation (plus ou moins précise) de la concentration de cet anticorps (peut demander de réaliser une gamme étalon au préalable)
 Méthode ELISA Direct ci-dessus (source en cliquant sur l'image)
Méthode ELISA Direct ci-dessus (source en cliquant sur l'image)
Définitions :
- Antigènes : toute substance reconnue comme étrangère par notre système immunitaire
- Anticorps : protéine capable de réagir avec un antigène
- Substrat : le plus souvent une substance solide support d'un réactif
- Epitope : Partie d'une molécule capable de stimuler la production d'un anticorps
Matériel :
- Plaques de microtitration 96 puits à fond plat
- Les différentes versions -
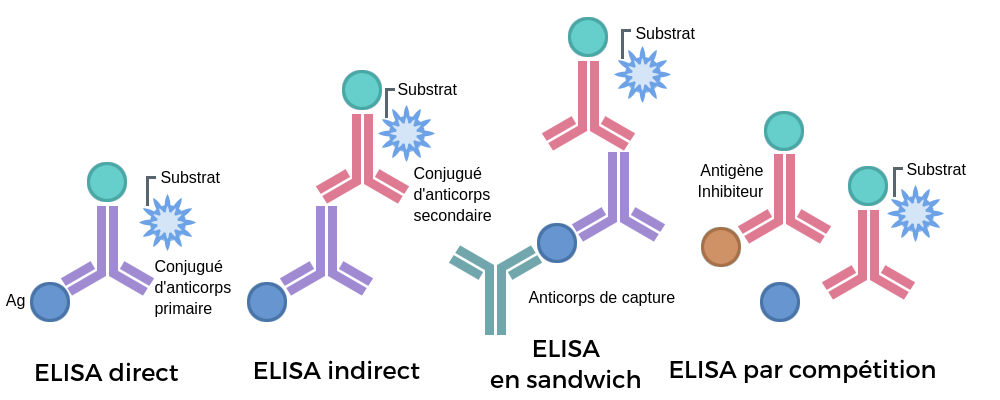
Il existe 4 versions différentes de la méthode ELISA en fonction de la précision recherchée et du contexte du test (approche quantitative ou qualitative).
-
ELISA Directe
Fonctionnement :
La méthode Direct n'utilise pas d'intermédiaire entre l'antigène et l'anticorps porteur de l'enzyme. On applique l'un sur l'autre puis on ajoute un "substrat chromogène", c'est à dire un révélateur qui permet de savoir si des antigènes ont été retenus.
Procédure :
- Fixer les antigènes (ou les anticorps selon le test souhaité). L'expérience s'effectue la plupart du temps sur des plaques de microtitration. Le fond des puits est recouvert d'un matériau capable d'absorber les antigènes déposés à sa surface, et donc de les fixer. On dit que les antigènes s'adsorbent.
- Laisser incuber la plaque permet de fixer durablement les antigènes dans le fond du puits.
- Rincer le puits afin de retirer l'excédent d'antigènes non fixés.
- Ajouter l'anticorps lié à l'enzyme capable de réagir avec le substrat. C'est à ce moment que des couples antigène-anticorps vont se former.
- Rincer le puits afin de retirer l'excédent d'anticorps non fixés à des antigènes.
- Ajouter le substrat chromogène permettant de déclencher la réaction lumineuse avec l'enzyme.
- Mesurer la concentration de l'anticorps en utilisant l'absorption de la solution.
-
ELISA Indirecte
Fonctionnement :
La méthode Indirecte utilise un intermédiaire en plus de l'antigène et de l'anticorps à doser. Un anticorps secondaire portant l'enzyme se fixe sur l'anticorps primaire fixé à l'antigène. Plusieurs anticorps secondaires peuvent se fixer à un anticorps primaire, augmentant ainsi la précision du dosage.
Procédure :
- Fixer les antigènes (ou les anticorps selon le test souhaité). L'expérience s'effectue la plupart du temps sur des plaques de microtitration. Le fond des puits est recouvert d'un matériau capable d'absorber les antigènes déposés à sa surface, et donc de les fixer. On dit que les antigènes s'adsorbent.
- Laisser incuber la plaque permet de fixer durablement les antigènes dans le fond du puits.
- Rincer le puits afin de retirer l'excédent d'antigènes non fixés.
- Ajouter l'anticorps primaire. C'est à ce moment que des couples antigène-anticorps vont se former.
- Laisser incuber la plaque permet de s'assurer qu'un maximum de liaisons antigènes-anticorps primaires se forment.
- Rincer le puits afin de retirer l'excédent d'antigènes primaires (et autres) non fixés.
- Ajouter l'anticorps secondaire lié à l'enzyme capable de réagir avec le substrat.
- Laisser incuber pour permettre la formation de complexes antigène-anticorps-enzyme.
- Rincer le puits afin de retirer l'excédent d'anticorps secondaires non fixés à des antigènes.
- Ajouter le substrat chromogène permettant de déclencher la réaction lumineuse avec l'enzyme.
- Mesurer la concentration de l'anticorps primaire en utilisant l'absorption de la solution.

-
ELISA Sandwich
Fonctionnement :
La méthode Sandwich implique l'utilisation de deux anticorps spécifiques. Le premier anticorps (capture) est fixé au fond du puits et capture l'antigène cible. Ensuite, un deuxième anticorps (détection) est ajouté, se liant à un autre épitope de l'antigène, formant ainsi un "sandwich" antigène-anticorps. L'anticorps de détection est généralement couplé à une enzyme qui, lorsqu'elle réagit avec un substrat, génère un signal détectable.
Procédure :
- Revêtir le fond des puits avec l'anticorps de capture spécifique à l'antigène cible.
- Incuber pour permettre la fixation de l'anticorps de capture.
- Rincer pour éliminer l'excès d'anticorps non fixé.
- Ajouter l'échantillon contenant l'antigène cible.
- Incuber pour permettre la formation du complexe antigène-anticorps.
- Rincer pour éliminer l'excès d'antigène non lié.
- Ajouter l'anticorps de détection spécifique à un autre épitope de l'antigène cible.
- Incuber pour permettre la formation du "sandwich" antigène-anticorps.
- Rincer pour éliminer l'excès d'anticorps de détection non lié.
- Ajouter le substrat chromogène pour déclencher la réaction enzymatique.
- Mesurer la réaction enzymatique pour estimer la concentration de l'antigène cible.

-
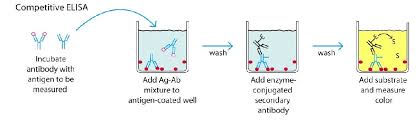
ELISA Competition
Fonctionnement :
La méthode de Compétition repose sur la compétition entre un antigène marqué et un antigène non marqué pour la liaison à un anticorps fixé au fond du puits. L'antigène de l'échantillon et l'antigène marqué (conjugué à une enzyme) entre en compétition pour se lier à l'anticorps spécifique fixé au fond du puits. La quantité d'antigène de l'échantillon est inversement proportionnelle au signal mesuré.
Procédure :
- Revêtir le fond des puits avec l'anticorps spécifique.
- Incuber pour permettre la fixation de l'anticorps.
- Rincer pour éliminer l'excès d'anticorps non fixé.
- Ajouter l'échantillon contenant l'antigène cible et l'antigène marqué (conjugué à une enzyme).
- Incuber pour permettre la compétition entre l'antigène de l'échantillon et l'antigène marqué pour se lier à l'anticorps.
- Rincer pour éliminer l'excès d'antigène et d'antigène marqué non lié.
- Ajouter le substrat chromogène pour déclencher la réaction enzymatique.
- Mesurer la réaction enzymatique pour estimer la concentration de l'antigène cible.
- Conclusion-
La méthode ELISA (Enzyme-Linked Immuno Assay) offre une approche polyvalente pour détecter la présence d'anticorps dans une solution et est largement utilisée dans divers domaines, notamment la recherche médicale, le diagnostic clinique et la surveillance des maladies infectieuses. Avec ses différentes versions, telles que ELISA Directe, ELISA Indirecte, ELISA Sandwich et ELISA Competition, elle permet une analyse précise et sensible, adaptée à une gamme étendue d'applications. En combinant des principes immuno-chimiques avec des réactions enzymatiques, l'ELISA fournit des résultats fiables et reproductibles, facilitant ainsi la prise de décision clinique et la recherche scientifique.
Sources :
Fiche Technique de l'ELISA
Définition
L'ELISA est une méthode biochimique permettant de détecter et de quantifier des substances telles que des peptides, des protéines, des anticorps et des hormones.
Principe
L'ELISA repose sur l'utilisation d'anticorps liés à une enzyme pour détecter la présence d'une substance cible (antigène) dans un échantillon. La réaction enzymatique produit un signal (souvent une couleur) qui est mesuré pour déterminer la quantité de cible présente.
Types d'ELISA
-
ELISA Direct: L'antigène est immobilisé sur une surface et détecté directement par un anticorps conjugué à une enzyme.
-
ELISA Indirect: L'antigène est immobilisé, puis détecté par un anticorps primaire non conjugué et un anticorps secondaire conjugué à une enzyme.
-
ELISA Sandwich: Utilise deux anticorps spécifiques à l'antigène, un pour la capture et un pour la détection.
-
ELISA Compétitif: L'échantillon et un antigène marqué avec une enzyme concourent pour la liaison à un anticorps spécifique.
Composants
-
Antigène: Molécule cible à détecter.
-
Anticorps: Protéines qui se lient spécifiquement à l'antigène.
-
Enzyme: Conjuguée à l'anticorps pour produire un signal détectable.
-
Substrat: Réagit avec l'enzyme pour produire un signal (souvent colorimétrique).
-
Plaques ELISA: Plaques à 96 puits généralement utilisées pour l'immobilisation des anticorps ou antigènes.
Étapes de l'ELISA
-
Coating: Immobilisation de l'antigène ou de l'anticorps sur la plaque.
-
Blocage: Saturation des sites non spécifiques avec une protéine bloquante pour éviter les liaisons non spécifiques.
-
Incubation avec l'échantillon: Ajout de l'échantillon contenant l'antigène ou l'anticorps cible.
-
Incubation avec l'anticorps conjugué à l'enzyme: Ajout de l'anticorps détecteur lié à une enzyme.
-
Lavage: Élimination des composants non liés.
-
Révélation: Ajout du substrat pour l'enzyme, entraînant une réaction colorimétrique.
-
Lecture: Mesure de l'absorbance à l'aide d'un spectrophotomètre.
Applications
-
Diagnostic médical: Détection de maladies infectieuses, de marqueurs tumoraux, de niveaux d'hormones.
-
Recherche: Quantification des protéines, des anticorps, des cytokines.
-
Industrie alimentaire: Détection de contaminants ou d'allergènes.
-
Vétérinaire: Diagnostic des maladies animales.
Schéma de l'ELISA
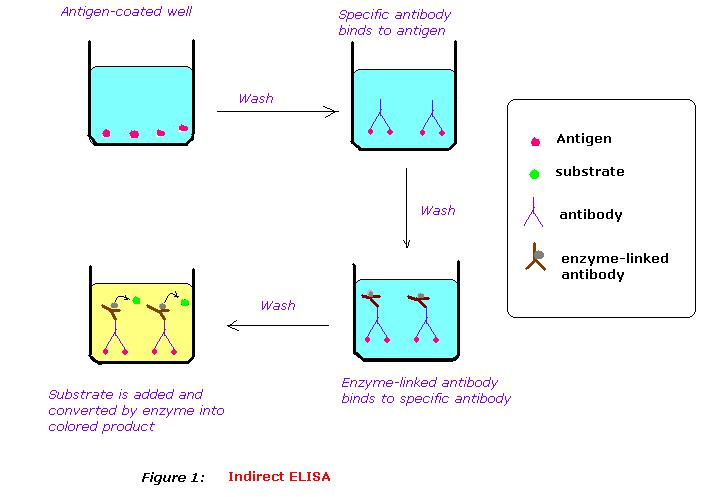
Tableau Résumé des Étapes de l'ELISA
|
Étape |
Description |
|---|---|
|
Coating |
Immobilisation de l'antigène ou de l'anticorps sur la plaque |
|
Blocage |
Saturation des sites non spécifiques |
|
Incubation avec l'échantillon |
Ajout de l'échantillon contenant l'antigène ou l'anticorps cible |
|
Incubation avec l'anticorps conjugué à l'enzyme |
Ajout de l'anticorps détecteur lié à une enzyme |
|
Lavage |
Élimination des composants non liés |
|
Révélation |
Ajout du substrat pour l'enzyme |
|
Lecture |
Mesure de l'absorbance avec un spectrophotomètre |
Optimisation de l'ELISA
-
Concentration des anticorps: Ajustement pour éviter la saturation ou une liaison insuffisante.
-
Conditions d'incubation: Optimisation du temps et de la température d'incubation.
-
Tampons de lavage: Utilisation de tampons appropriés pour minimiser les liaisons non spécifiques.
-
Contrôle négatif et positif: Inclusion pour valider les résultats.
Variantes de l'ELISA
-
ELISA Direct: Simplicité et rapidité, mais risque de bruit de fond élevé.
-
ELISA Indirect: Sensibilité accrue grâce à l'amplification du signal.
-
ELISA Sandwich: Très spécifique et sensible, idéal pour des échantillons complexes.
-
ELISA Compétitif: Utile pour les petits antigènes ou lorsque la quantification directe est difficile.
Liens et Articles Scientifiques
-
Article de base sur l'ELISA: Engvall et Perlmann, 1971
-
Optimisation des conditions de l'ELISA: Crowther, 2000
-
Utilisation de l'ELISA en diagnostic: Schüpbach et al., 1984
-
ELISA pour la détection des anticorps: Van Weemen et Schuurs, 1971
-
Avancées récentes dans l'ELISA: Lequin, 2005
Séquençage de l'ADN
-
C'est quoi un ADN :
L'ADN, ou acide désoxyribonucléique, est une molécule présente dans toutes les cellules vivantes. C'est une macromolécule biologique qui contient les instructions génétiques essentielles nécessaires au développement, au fonctionnement, à la croissance et à la reproduction de tous les organismes vivants, qu'ils soient simples ou complexes.
L'ADN se présente sous la forme d'une double hélice, semblable à une échelle enroulée, composée de deux brins complémentaires d'unités de base appelées nucléotides. Chaque nucléotide se compose d'un sucre (désoxyribose), d'un groupement phosphate et d'une base azotée. Les quatre bases azotées trouvées dans l'ADN sont l'adénine (A), la thymine (T), la cytosine (C) et la guanine (G). Ces bases se lient spécifiquement l'une à l'autre, avec l'adénine s'associant à la thymine et la cytosine s'associant à la guanine, formant ainsi les "barreaux" de l'échelle de la double hélice.
L'ADN contient les informations génétiques codées sous forme de séquences de nucléotides. Ces informations sont transmises d'une génération à l'autre et sont utilisées par les cellules pour fabriquer des protéines, qui sont les principaux acteurs de nombreuses fonctions biologiques dans les organismes vivants.

Cliquez sur la photo pour voir une vidéo explicative
-
Définition du séquençage d'ADN:
Le séquençage de l'ADN (acide désoxyribonucléique) consiste à déterminer la succession linéaire des bases A, C, G et T qui composent la structure de l'ADN. Il est intéressant d'avoir la séquence de l'ADN car on a accès à toutes les informations qui la contiennent (Lamoril, J. et al. 2008).
-
Applications
Le séquençage est une technique essentielle avec diverses applications. En médecine, il est utilisé pour diagnostiquer les maladies génétiques et les mutations associées aux cancers, ce qui guide les traitements. En recherche génétique, il aide à comprendre la structure et la fonction des gènes. La pharmacogénomique exploite les variations génétiques pour personnaliser les traitements. Les microbiomes et la génomique environnementale révèlent la diversité génétique des organismes et leur interaction avec leur environnement. Le séquençage de novo permet d'étudier des organismes sans séquence de référence. L'évolution est étudiée en comparant les séquences génétiques entre espèces ou populations. En agronomie, il est utilisé pour améliorer les plantes et les animaux d'élevage. Ces applications illustrent la polyvalence du séquençage dans divers domaines de recherche et de médecine.
-
Historique
Le séquençage de l'ADN est inventé dans la deuxième moitié des années 1970. Deux méthodes sont développées indépendamment, l'une par l'équipe de Walter Gilbert, aux États-Unis, et l'autre par celle de Frederick Sanger (en 1977), au Royaume-Uni, Pour cette découverte, Gilbert et Sanger sont récompensés par le prix Nobel de chimie en 1980.
-
Techniques
Les deux premières techniques sont celle de Maxam-Gilbert et celle de Sanger, Elles étaient décrites en 1977, Il existe aussi d’autres techniques modernes.
Voici quelques techniques avec leurs méthode de fonctionnement :
- Technique de Maxam-Gilbert: C’est une méthode chimique de séquençage,Les réactifs clivent spécifiquement après chacune des bases A, C, G, [A + G], [C + T]. Cette technique est basée sur la propriété de certains agents chimiques, l’hydrazine, le diméthyl sulfate (DMS) et l’acide formique, de modifier les bases de l’ADN. Dans un second temps, la pipéridine est ajoutée et « casse » les brins d’ADN au niveau des bases modifiées (Figure-1) . Les agents chimiques sont utilisés dans des conditions telles qu’ils n’agissent qu’avec un faible pourcentage des bases de l’ADN étudié. L’ADN à séquencer est marqué à une extrémité. Le plus souvent, il s’agit d’un marqueur radioactif. Le produit de séquence est déposé sur un gel d’acrylamide, puis la séquence lue après autoradiographie (Figure-2). L’ADN étudié peut être simple ou double brin. Cette technique permettait d’analyser des fragments allant jusqu’à 500 pb.

Figure (1)

Figure (2)
· Technique de Sanger: Elle a en effet rapidement dépassé la méthode de Maxam-Gilbert pour la remplacer et reste à ce jour la principale méthode de séquençage utilisée dans les laboratoires. Cependant, elle reste limiter car elle ne peut sequencer que des fragments entre 400-800pb. Son principe est le suivant.
-
Préparation de l'ADN: Tout d'abord, l'ADN à séquencer est préparé. Il peut s'agir d'un fragment d'ADN spécifique ou d'une séquence plus longue.
-
PCR : L'ADN est amplifié par PCR pour obtenir suffisamment de matériel génétique pour l'analyse. Puis, il faudra dénaturer les échantillons obtenus pour obtenir un ADN simple brin.
- Réaction de séquençage: A partir de notre ADN, on obtient un ADNc marqué grâce au mélange réactionnel qui contient les tampons, l’ADN polymérase, des déoxynucléotides triphosphates (dNTP, dA-, dC-, dG-, dT-TP) mais aussi des didéoxynucléotides triphosphates (ddNTP, ddA-, ddC-,ddG-, ddT-TP) couplés à un fluorophore différents. Lors de cette réaction, il y a statistiquement au moins une chance pour chaque base qu'un ddNTP soit attribué à la place d'un dNTP, et arrêtera ainsi l'action de l'ADN polymérase. Nous obtiendrons alors des échantillons de taille différentes. (Figure 3)
- Électrophorèse: De nos jours, il faut utiliser un automate de séquençage pour effectuer une électrophorèse de capillaire (sinon le travail est fastidieux).
-
Analyse des données: Les données de séquençage sont ensuite analysées par ordinateur pour assembler la séquence complète et déterminer l'ordre précis des nucléotides dans l'ADN.

Figure (3)
· Séquençage par hybridation :
Le séquençage par hybridation repose sur l’utilisation de puces à ADN contenant de plusieurs centaines (pour les puces de première génération) à plusieurs milliers d’oligonucléotides (courtes chaînes de nucléotides) . L’ADN à analyser est coupé en de multiples fragments qui sont ensuite incubés sur la puce où ils vont s’hybrider avec les oligonucléotides dont ils sont complémentaires. La lecture de la puce (la détection des oligonucléotides hybridés), permet d’obtenir le spectre de la séquence d’ADN, c’est-à-dire sa composition en sous-séquences de n nucléotides, où n est la taille des sondes sur la puce utilisée. Le traitement informatique du spectre permet ensuite de reconstituer la séquence entière. (Figure-4)

Figure (4)
· Technique Shotgun :
La technique de Shotgun sequencing consiste brièvement à fragmenter l'ADN en petits morceaux, à séquencer chaque fragment individuellement, puis à assembler les séquences pour reconstruire le génome ou le fragment d'ADN d'origine.
· Principe de séquençage de grands génomes:
L’automatisation à l’aide de robots a permis de séquencer de nombreux génomes dont le génome humain. Pour réaliser le séquençage d’un grand génome, une carte physique du génome est d’abord constituée.
· Amplification génome complet / whole genome amplification (WGA) :
La WGA est une méthode qui permet d'amplifier l'ensemble du génome à partir d'un échantillon biologique, fournissant ainsi suffisamment d'ADN pour les analyses ultérieures, même à partir d'échantillons de faible quantité ou de mauvaise qualité.
· Pyroséquençage :
Le pyroséquençage est une méthode basée sur la détection de la libération de pyrophosphate qui a lieu lors de l'incorporation de nucléotides. Le brin d'ADN doit d'abord être amplifié par PCR avant le séquençage. L'ordre dans lequel les nucléotides seront ajoutés est ensuite choisi dans le séquenceur (c'est-à-dire G-A-T-C). Chaque fois qu'un nucléotide spécifique est ajouté dans la chaîne par l'ADN polymérase, un pyrophosphate est libéré et converti en ATP par une ATP sulfurylase. L'ATP stimule alors l'oxydation de la luciférine en luciférase, cette réaction génère un signal lumineux enregistré sous la forme d'un pic. L'incorporation de chaque nucléotide est alors corrélée à un signal. Le signal lumineux est proportionnel à la quantité de nucléotides incorporés lors de la synthèse du brin d'ADN (deux nucléotides incorporés correspondent à deux pics). Lorsque les nucléotides ajoutés ne sont pas incorporés dans la molécule d'ADN, aucun signal n'est enregistré. Cette technique ne nécessite ni nucléotides marqués par fluorescence ni électrophorèse sur gel.
· Séquençage par spectrométrie de masse :
La spectrométrie de masse pour le séquençage fragmente l'ADN, ionise les fragments, analyse leur masse pour reconstruire la séquence. Bien qu'alternative, elle est limitée en taille de fragments et précision, souvent combinée à d'autres méthodes pour des résultats plus fiables.
- Séquençage par nanopore :
Le séquençage de l'ADN par les nanopores est une méthode utilisée depuis 1995. Elle permet de déterminer l'ordre dans lequel les nucléotides sont disposés sur un fragment d'ADN donné à l'aide de nanopores (trou avec un diamètre de l'ordre du nanomètre).
Des protéines cellulaires transmembranaires possédant des petits pores peuvent agir comme des nanopores. Des nanopores peuvent également être construits dans un résidu de silicium simplement en y effectuant un trou d'une dizaine de nanomètres. Le morceau de silicium est par la suite rempli lentement par sculptage à l'aide de faisceau d'ions.
La méthode nanopore est une technique de séquençage d'ADN basée sur l'utilisation de nanopores, de minuscules trous à l'échelle nanométrique. Méthodologie:
-
-
Préparation de l'échantillon: L'ADN à séquencer est préparé et dénaturé pour obtenir des brins simples.
-
Passage à travers le nanopore: Les brins d'ADN sont acheminés à travers un nanopore intégré dans une membrane, généralement en silicium ou en polymère. Un potentiel électrique est appliqué à travers la membrane, ce qui entraîne le passage des ions à travers le nanopore.
-
Mesure de la conductance ionique: Pendant que l'ADN traverse le nanopore, il perturbe le flux d'ions à travers celui-ci. Cette perturbation est mesurée sous forme de variations de conductance ionique, ce qui permet de déduire des informations sur la séquence d'ADN.
-
Analyse des signaux: Les variations de conductance ionique sont enregistrées et analysées pour reconstruire la séquence d'ADN. Chaque base nucléotidique produit une signature caractéristique de conductance ionique, permettant ainsi l'identification de la séquence.
-
La méthode nanopore présente plusieurs avantages, notamment sa rapidité, sa simplicité et sa capacité à séquencer de longues séquences d'ADN sans nécessiter d'amplification préalable. Elle est utilisée dans divers domaines de la recherche en génétique, en médecine et en biologie, ainsi que dans des applications de diagnostic et de surveillance environnementale.
-
Conclusion:
Le séquençage de l’ADN par la méthode de Sanger est actuellement la méthode de choix dans les laboratoires hospitaliers et de recherche . Sans cesse améliorée depuis plus d’une dizaine d’années, elle semble atteindre aujourd’hui ses limites même si des améliorations, notamment la miniaturisation de cette technique sont en développement . Outre les limites technologiques, le coût du séquençage selon cette méthode est encore élevé .
La méthode nanopore commercialisée à partir de 2016 est aussi souvent utilisée vu les avantages qu'elle offre.
-
Bibliographie :
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7147846/
https://fr.wikipedia.org/wiki/S%C3%A9quen%C3%A7age_de_l%27ADN#
https://fr.wikipedia.org/wiki/S%C3%A9quen%C3%A7age_par_nanopores#
Electrophorèse
I - Introduction :
L’électrophorèse - technique de séparation des molécules chargées sous l'effet d'un champ électrique appliqué par deux électrodes (négative et positive)
L'électrophorèse sur gel est une technique de laboratoire utilisée en biologie moléculaire et biochimie pour séparer des extraits contenant de l’ADN, de l’ARN et des protéines en fonction de leur taille et de leur poids moléculaire.
Matériel utilisé :
- Cuve d'électrophorèse avec un peigne (pour l'électrophorèse horizontal)
- Kits Western Blot (pour l'électrophorèse verticale) voir lien
- Tampon de migration
- Agarose ou polyacrylamide
- Balance de précision
- ErlenMeyer
- Marqueur de poids moléculaires (DNA Ladder)
- Tampon de charge
- UV (visualisation)
II - Électrophorèse horizontale de l'ADN sur gel d'agarose
L’électrophorèse horizontale sur gel est une méthode utilisée dans la séparation de l’ADN. Une extrémité contient une anode et l’autre une cathode. L'ADN, en étant chargé négativement, migre vers l’électrode positif selon sa taille et son poids moléculaire.
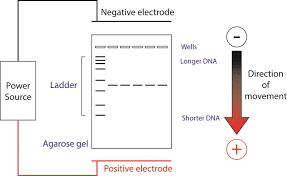
Figure 1 : principe de l'électrophorèse horizontale
Préparation d’un gel d’agarose 1%
1. Pesez 1 g d'agarose et mettez dans l’Erlenmeyer-150ml. Ajoutez 100 ml de tampon (TBE) (on peut placer le 2ème ErlenMeyer-25ml comme un bouchon dans l’ErlenMeyer-150ml)
2. Chauffez au four micro-ondes jusqu’à la dissolution complète de l’agarose. Mélangez de temps en temps.
ATTENTION : Très chaud ! Portez des gants de protection !
3. Laissez reposer le gel à température ambiante durant 10 min.
4. Assemblez le plateau avec un peigne de 19 puits et coulez le gel.
5. Ajoutez de l'intercalant de l'ADN (par exemple Atlas ClearSight DNA Stain) et mélangez.
ATTENTION : L'intercalant de l'ADN est un composé toxique ! Portez des gants !
6. Coulez le gel et laissez polymériser au moins 30 min.
7. Ajoutez le tampon de charge (TpC)
8. Retirez le peigne du gel, placez le plateau avec le gel dans la cuve de migration. Mettez le tampon de migration (par exemple TBE-0.5x) dans la cuve pour couvrir le gel.
9. Déposez les échantillons sur le gel.
10. Lancez la migration : 100 V, 60 min.
11. Observez l’ADN sous la lampe à UV
12. Utilisez le DNA Ladder afin de repérer la quantité relative de l'ADN

Figure 2 : exemple du DNA Ladder (1 kb) - marquer du poids moléculaire


Figure 3 : cuve pour électrophorèse avec un peigne de 19 puits (ouverte - gauche; fermée - droite)
III - Électrophorèse verticale des protéines sur gel de polyacrylamide SDS-PAGE
L'électrophorèse des protéines sert à la quantification relative des protéines à partir d'un extrait suite à la séparation des protéines sur gel. La séparation s'effectue en fonction de la masse moléculaire. On peut effectuer l'électrophorèse PAGE en conditions non-dénaturantes (Poly-Acrylamide Gel Electrophoresis) mais également SDS-PAGE en conditions dénaturantes (Sodium Dodecylsulfate-PAGE).
Pour SDS-PAGE : Les protéines sont dénaturées dans un tampon de charge (tampon laemmli) contenant du
SDS (charge négativement les protéines en les déroulant) et un réducteur (β‐mercaptoéthanol ou
DTT) qui détruit les ponts disulfures. Les protéines, alors chargées négativement, migrent dans le
gel de polyacrylamide sous l'effet du champ électrique, en fonction de leur poids moléculaire apparent
*On pourrait éventuellement utiliser le gel d'agarose mais en raison de la taille plus petite des pores du gel de polyacrylamide, une séparation plus précise peut être obtenue
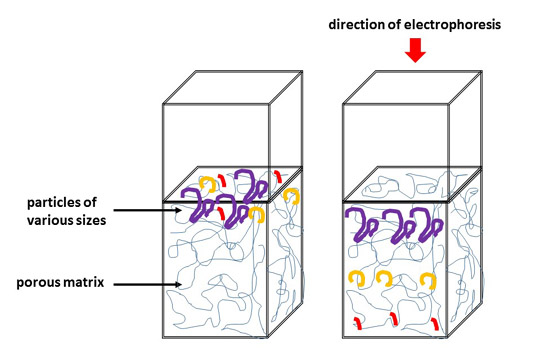
Figure 4 : principe de l'électrophorèse verticale
Les étapes principales
- Préparation des échantillons. Dénaturation des protéines par SDS et la réduction des ponts disulfures par un réducteur.
- Préparation de la cuve d’électrophorèse et du gel (Adapter la cassette de gel dans son support)
- Remplissage de la cuve. Remplir le compartiment intérieur (entre la cassette de gel et la plaque de barrage) et le compartiment principal avec du tampon de migration
- Dépôt des échantillons sur le gel
- Mise en route de l’électrophorèse

Figure 5 : Exemple de la migration des protéines sur gel
Cas pratique : Fabrication d'un gel de polyacrylamide et migration de protéines
-
Préparation d'un gel de polyacrylamide (1H30) :
Pour un gel de 0,75mm d'épaisseur
Matériel :
- Lames 0,75mm
- Support à lames
- Propipettes
- Tubes falcon ou bécher
- Glace
- Éprouvette graduée
- Pipette pasteur
- Peigne adapté à la largeur des lames
| SOLUTIONS MÈRES |
GEL DE SÉPARATION |
GEL DE CONCENTRATION |
| (1) Acrylamide/bis-acryl 40% |
1,9 mL (7,5%) |
0,5 mL (4,0%) |
| (2) Tris-HCL 1,5M pH 8,8 |
2,5 mL (0,375M) |
X |
| (3) Tris-HCL 0,5M pH 6,8 | X |
1,25 mL (0,125M) |
| (4) SDS 10% |
100 µL (0,1%) |
50 µL (0,1%) |
| (5) H2O distillée |
5,4 mL |
3,15 mL |
| (6) APS 10% |
100 µL (0,1%) |
50 µL (0,1%) |
| (7) TEMED |
10 µL |
5 µL |
Protocole :
- Préparer les deux gels séparément en introduisant dans un tube falcon les réactifs (1), (2), (4) et (5) pour le gel de séparation et les réactifs (1), (3), (4) et (5) dans un autre tube pour le gel de concentration.
- Préparer les lames en les insérant dans leur support en en faisant une marque à 5cm de hauteur
- Vérifier l'étanchéité du montage en introduisant de l'eau distillée, puis vider et sécher
- Mettre le tube du gel de séparation dans la glace (pour ralentir la polymérisation) et y ajouter le TEMED et l'APS
- Mélanger délicatement en retournant le tube (!! Ne pas agiter pour éviter qu'il ne se forme trop de bulles !!).
- Remplir de gel de séparation à hauteur de la marque en versant doucement sur les bords (tapoter pour équilibrer le niveau et faire remonter de potentielles bulles)
- Ajouter très délicatement une fine couche d'isopropanol ou d'eau distillée
- Laisser prendre pendant 30min (+ ou -)
- Bien rincer délicatement à l'eau distillée et égoutter
- Placer le tube de gel de concentration dans la glace et ajouter TEMED et APS
- Couler le gel de la même manière que le premier jusqu'à ras bord.
- Insérer le peigne et remuer jusqu'à ce qu'il n'y ait plus de bulles ( !! Bien porter des lunettes pour se protéger dans projections !! )
- Attendre 30 min
Ne pas détacher les plaques de verre avant la fin de la migration
-
Migration de protéines
Préparation du tampon de migration
Notre tampon était déjà préparé en 5X (qu'on a dilué en 1X). Pour autant, voici sa composition :
| SOLUTIONS MÈRES | TAMPON DE MIGRATION 1X |
| Tris / Glycine 10X |
300 mL (1X) |
| SDS 20% | 4 mL (0,1%) |
| Eau distillée |
3L |
Migration
La première étape consiste à préparer la cuve :
- Placer le gel sur un support. Si il y a plus d'emplacement que de gel par support, bien prévoir d'utiliser un "fantôme" pour assurer la conduction du courant.
- Remplir la cuve au tiers de son volume de tampon 1X et attendre quelques minutes que le gel en absorbe.
- Continuer à remplir la cuve progressivement en laissant reposer le chaque fois. S'arrêter un fois que la hauteur des puits est dépassée (y aller doucement pour éviter les bulles) mais ne pas monter trop haut.
Il faut ensuite centrifuger le marqueur et les échantillons puis les déposer en alternance dans les puits (on commence au bord par la marqueur, puis protéines, marqueur...)
Enfin, brancher la cuve au générateur (120V) et faire migrer pour environ 1H
Isolation de l'ADN de plantes deshydratées
Des techniques utilisées précédemment consistaient a lyophiliser les plantes avant extraction.
Avantages
Dans ce processus de lyophilisation il n'est pas nécessaire de laisser le tissu dans le congélateur. Cette technique nous permet de pouvoir facilement transporter et traiter mécaniquement le tissu.
Inconvénients
Les lyophilisateurs sont relativement chères et ont une capacité limité. La lyophilisation nécessite plusieurs jours. Des erreurs dans l'opération peuvent causer la dégradation de l'ADN.
Méthode d'Isolation
Il existe une autre méthode qui consiste a déshydrater les tissus de plantes et traiter et extraire l'ADN ayant une masse moléculaire élevée. Elle peut être utilisé sur une grande variété de plantes et sur un grand nombre d'échantillons. Cette technique permet d'obtenir un rendement entre 70 à 150 mg d'ADN totale par gramme de tissue hydraté. Quand le tissu est déshydraté il peut être préservé et rester stable pendant plusieurs jours sans perte de rendement ou de qualité. Il est aussi plus facile a manipuler.
Matériel
- Déshydrateur alimentaire
- Moulin à café
- Billes de verre
- Mélangeur de peinture
- Petites pochettes en tissue 7,6cm x 12,7 cm
Tampon d'Extraction
- 100 mM Tris-HCl pH 8
- 50 mM EDTA pH 8
- 500 mM NaCl
- 1.25% SDS (w/v)
- 0.38 g sodium bisulfite par 100 mL (tampon ajouté avant utilisation = TE tampon)
- 50 mM Tris-HCl pH 8
- 10 mM EDTA
- acétate de potassium 5M
Méthodes
Prétraitement
-
- Collecter les tissues de feuilles (3 à 5g poids avant séchage)
- Les placer dans des sachets fermés puis dans le déshydrateur et sécher à 45-55°C pendant 12 à 24 heures.
- Broyer le tissu sec et placer dans un tube de centrifugation de 50mL de polypropylène contenant des billes de verre (10 g).
- Mélanger avec un mixeur de peinture jusqu'à obtenir une poudre fine (30s à 3min)
- Les billes de verre sont laissées dans le tube pendant la première centrifugation et ils sont éliminées avec le culot de centrifugation ou ils sont enlevées et rincées pour une réutilisation futur.
- Les tissus fibreux (riz...) sont broyés avec un moulin a café (20 à 40s) puis transféré dans un tube de 50mL
- Après le prétraitement des tissus des plantes on commence l'isolation (d’après Dellaporta et al. (1983))
Isolation
- Ajouter 16 mL de tampon d'extraction (chauffer à 65°) au tissu poudré. Mélanger avec une spatule ou inversant le tube et incuber a 65° pendant 10 à 15 min.
- Ajouter 5 mL d'acétate de potassium à 5M. Bien mélanger et incuber sur glace pendant 20 min.
- Centrifuger a 3000 rpm pendant 20 min. Verser le surnageant à travers un filtre Miracloth dans un autre tube de 50 mL.
- Ajouter 2/3 vol. d'isopropanol ou 2 vol éthanol et incuber les tubes à 20°C pendant 30 min pour précipiter l'ADN.
- Centrifuger à 3000 rpm pendant 5 à 10 min. Laver avec 70% d'éthanol et sécher à l'air pendant 10 à 15 min.
- Suspendre le culot d'ADN dans 500 a 1000 µL de tampon TE. Transférer dans un tube de 5mL et centrifuger à 3000 rpm pendant 15 à 20 min. Transférer le surnageant dans un nouveau tube de 5mL.
- Ajouter 10µL de RNAase (10 mg/mL) a l'échantillon et incuber à température ambiante pendant 10 à 15 min. Ajouter 1/10 vol. 3 M d'acétate de sodium et 2 vol 95% éthanol puis incuber à -20°C pendant 30 min pour précipiter l'ADN.
- Centrifuger le culot d'ADN à 3000 rpm pendant 5 à 10 min. Laver avec 70% éthanol, sécher, et suspendre dans 1/2 vol. de tampon TE dans un tube de 1.5 mL. Centrifuger à 10000 rpm pendant 15 à 20 min. Transférer le surnageant dans un nouveau tube de 1.5 mL.
Extraction d'ADN de cellules végétales
En biologie moléculaire, de nombreuses techniques se basent sur l'analyse et le traitement de l'ADN (Acide DesoxyriboNucléique), à ne pas confondre avec l'ARN (Acide RiboNucléique).
C'est le cas de la Polymérase Chain Reaction (PCR), Southern Blot, utilisation d'enzymes de restriction, hybridation moléculaire...
Pour ce faire, il est nécessaire d'isoler l'ADN de son environnement (cellule, tissus) pour obtenir d'un échantillon pur et et quantité suffisante pour servir de base pour les techniques suivantes.
Protocole d'extraction : [1]
- Couper une pièce de feuille (1-2cm2) dans un tube eppendorf
- Broyer les feuilles fortement pendant 15 secondes.
- Ajouter 500 μL de tampons d’extraction
- Vortex 5 secondes.
- Centrifuger l’extrait pendant 5min a 13'000 rpm (4°C)
- Prélever 350 μl du surnageant dans un nouveau tube eppendorf
- Ajouter 350 μl d’isopropanol
- Inverser le tube une dizaine de fois et laissetr précipiter l’ADN pendant 2 min
- Centrifuger pendant 15 min à 13'000 rpm (4°C)
- Jeter le surnageant, ajouter 400 μl d’ethanol 80%.
- Centrifuger pendant 5 min à 13'000 rpm
- Enlever le surnageant et laisser sécher le culot (10min environ)
- Ajouter 50 μl d’ H2O distillé
- Vortex
L'isopropanol provoque l'agrégation et la précipitation de l'ADN dans la solution.
Le Vortex est un matériel en biologie moléculaire pour mélanger des solutions notamment dans des microtubes. Il est composé d'un socle lourd ou se situe le moteur de l'appareil. Au-dessus, se trouve un réceptacle en caoutchouc ou l'on pose le tube a votexer. [2]
Précisions :
Le tampon d'extraction est habituellement préfabriqué, il sert à rompre les membranes cellulaires et nucléaires, maintenir un pH optimal tout au long de l’extraction n, sauvegarder l'intégrité de l'ADN et prévenir sa dégradation, Séparer l'ADN des débris cellulaires et des contaminants. [2]
Protocole de fabrication du tampon d'extraction de cellules végétales : [3]
- Tampon d'extraction CTAB (pH 8)
- Tris-Cl 100 mM, pH 8,0
- EDTA 20 mM, pH 8,0
- NaCl 1,4 M
- 2 % (p/v) CTAB (bromure de cétyltriméthylammonium)
- 1 % PVP 40 000 (polyvinylpyrrolidone)
- 0,3 % de 2-β-mercaptoéthanol (ajouté directement avant utilisation dans une hotte)
- Chloroforme : alcool isoamylique (24:1 v/v)
- NaCl 6 M
- Acétate de potassium 3 M
- Glace froide
- Alcool isopropylique à 100 %
- Éthanol à 70 %
- Réhydratation dans un tampon TE 1 × (Tris-HCl 10 mM, pH 8,0 ; EDTA 1 mM, pH 8,0, autoclavé)
Bibliographie :
[1] Extraction d'ADN génomique et amplification des gènes d'actine 2 et Cycline D des plantes Arabidopsis Thaliana et Tomates, Laboratoire de Physiologie Végétale,Université de Neuchâtel : https://www.unine.ch/files/live/sites/physiologievegetale/files/shared/documents/TP-PCR.pdf
[2] Vortex, Wikipédia : https://fr.wikipedia.org/wiki/Vortex_(biologie_mol%C3%A9culaire)
[3] Fabriquer et utiliser des tampons d'extraction ADN courants, Fourni-Labo : https://www.fourni-labo.fr/tutoriel/comment-fabriquer-et-utiliser-des-tampons-d-39-extraction-d-39-adn-courants-2023-08-19
Dosage des chlorophylles
Le dosage des chlorophylles A et B est une expérience simple et basique de biologie végétale, par exemple lorsque l'on étudie l'effet d'un paramètre du milieu (carence en fer, exposition, etc).
Nous profitons de l'écriture du wiki à propos du mélangeur/disperseur turrax pour donner un exemple d'utilisation ici.
Protocole :
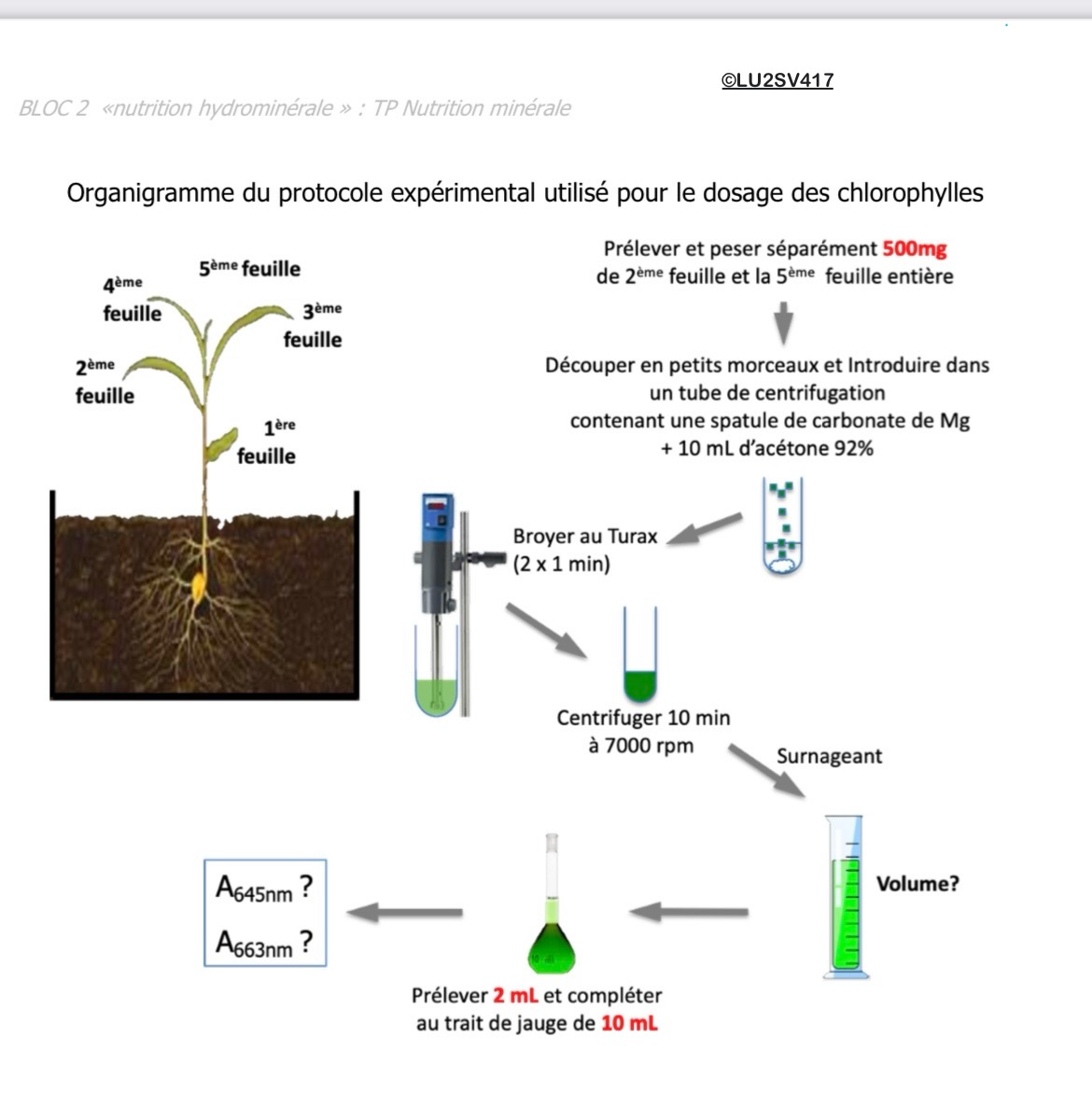
Nous cherchons à connaître la teneur en chlorophylle a et b de ces deux feuilles de tabac, l'une (celle de gauche) étant un peu plus âgée que l'autre :



Mesure de la masse pour ramener la concentration en chlorophylle de l'échantillon (mg.L-1) à la teneur dans la feuille (mg.g-1). (Mettre une coupelle pas comme sur les photos)



Débrancher l'appareil lors de la mise en place sur la potence. Porter des lunettes de protection, une blouse et s'attacher les cheveux.


La loi de beer lambert pour les deux longueurs d'onde, les concentrations des chlorophylles a et b ainsi que leur coefficient d'absorption spécifique nous permettes de poser le système d'équations ci-dessous :
Chla = 12,7 x A663 - 2,63 x A645 en mg.L-1
Chlb = 22,9 x A645 - 4,68 x A663 en mg.L-1
La teneur (mg de chlorophylle/g de feuille) est donnée par (concetration x dilution x volume extrait)/masse feuille
| A645 | A663 | Concentration Chla | Concentration Chlb | masse | teneur Chla | teneur Chlb | Chla+Chlb | |
| feuille 1 | 0,176 | 0,385 | 4,42662 mg.L-1 | 2,2286 mg.L-1 | 1,4895g | 0,294mg/g | 0,150mg/g | 0,446mg/g |
| feuille 2 | 0,283 | 0,635 | 7,32021 mg.L-1 | 3,5089 mg.L-1 | 1,5796g | 0,463mg/g | 0,222mg/g | 0,685mg/g |
Tampons d'extraction
Introduction
Un étape importante dans l’analyse de protéines ou d'ADN, est l'extraction de ces derniers des tissus et la purification des solution obtenus. Ces étapes ont lieu dans une solution tampon (solution qui maintient un pH à peu près stable, malgré certains changements des facteurs externes ou internes).
En sélectionnant les solutions appropriées et en suivant la méthode adéquate pour extraire l'ADN, on pourrait :
I) Les étapes générales de l'utilisation des tampons d'extraction d'ADN
1. Préparation des échantillons
- Collecter les échantillons biologiques contenant l'ADN que vous souhaitez extraire. Cela peut être du sang, des tissus, des cellules, etc.
- Préparer les échantillons en les broyant ou en les homogénéisant pour libérer les cellules et l'ADN à extraire.
2. Préparation des tampons
- Préparer les tampons d'extraction d'ADN nécessaires en suivant les protocoles spécifiques pour chaque type de tampon. Assurez-vous de respecter les concentrations et les conditions de pH recommandées.
3. Lyse cellulaire
- Ajouter le tampon de lyse à vos échantillons pour rompre les membranes cellulaires et nucléaires et libérer l'ADN. Les tampons de lyse contiennent souvent des agents détergents tels que le Triton X100, le SDS ou le CTAB pour dissoudre les membranes.
4. Inactivation des enzymes
- Si nécessaire, ajouter des agents inhibiteurs d'enzymes tels que l'EDTA pour empêcher la dégradation de l'ADN par des enzymes telles que les DNases ou les RNases.
5. Précipitation de l'ADN
- Ajouter des agents de précipitation ou des solutions de précipitation, tels que l'alcool isopropanol ou l'éthanol, pour précipiter l'ADN des solutions.
6. Centrifugation et lavage
- Centrifuger les échantillons pour séparer l'ADN précipité des autres composants cellulaires et des contaminants.
- Laver l'ADN précipité avec de l'éthanol pour éliminer les résidus de tampon et d'autres contaminants.
7. Réhydratation de l'ADN
- Dissolver l'ADN précipité dans un tampon d'hydratation approprié, tel que TE (Tris-EDTA), pour le stockage ou pour le préparer à des analyses ultérieures.
8. Analyse de l'ADN
- L'ADN extrait peut être utilisé pour diverses applications, telles que la PCR (réaction de polymérisation en chaîne), le séquençage, l'électrophorèse sur gel, etc.
Il est important de suivre attentivement les protocoles spécifiques à chaque kit d'extraction ou à chaque méthode d'extraction d'ADN pour obtenir les meilleurs résultats.
II) Les tampons les plus courants en Biologie
1) Tampon de chlorure de sodium (NaCl) :
- Principe : Le tampon de NaCl est souvent utilisé pour ajuster la force ionique des solutions. Il est généralement préparé en dissolvant du chlorure de sodium dans de l'eau distillée.
- Exemples d'utilisation : Extraction d'ADN, de protéines ou d'autres biomolécules, hybridation d'acides nucléiques, réactions enzymatiques.
- Préparation :
1. Dissoudre le chlorure de sodium dans de l'eau distillée pour obtenir la concentration désirée.
2. Stériliser par autoclavage si nécessaire.
- Utilisation :
1. Ajuster la force ionique des tampons ou des solutions de lavage dans diverses applications biologiques.
2. Utiliser comme tampon pour l'extraction d'ADN ou de protéines.
2) Tampon d'acétate de sodium :
- Principe : Le tampon d'acétate de sodium est utilisé pour maintenir un pH stable dans une plage légèrement acide (pH autour de 5). Il est composé d'acide acétique et de sel de sodium.
- Exemples d'utilisation : Extraction d'ADN plasmidique, purification de protéines par précipitation, électrophorèse d'acides nucléiques.
- Préparation :
1. Dissoudre l'acide acétique et le sel de sodium dans de l'eau distillée.
2. Ajuster le pH avec de l'acide acétique ou de l'hydroxyde de sodium jusqu'à obtenir le pH désiré.
- Utilisation :
1. Utiliser comme tampon de précipitation pour la purification de protéines.
2. Ajuster le pH des solutions pour différentes applications en biochimie.
3) Tampon de phosphate d'ammonium :
- Principe : Ce tampon est utilisé pour maintenir un pH stable dans une plage légèrement acide (pH autour de 5 à 6). Il contient des sels de phosphate et d'ammonium.
- Exemples d'utilisation : Extraction d'ADN, électrophorèse d'acides nucléiques, réactions enzymatiques.
- Préparation :
1. Dissoudre les sels de phosphate et d'ammonium dans de l'eau distillée.
2. Ajuster le pH avec de l'acide phosphorique ou de l'ammonium hydroxyde jusqu'à obtenir le pH désiré.
- Utilisation :
1. Utiliser comme tampon pour l'extraction d'ADN ou d'ARN.
2. Ajuster le pH des solutions pour différentes applications en biologie moléculaire.
4) Tampon de glycérine :
- Principe : La glycérine est souvent utilisée comme additif dans les tampons pour améliorer la stabilité des échantillons biologiques, réduire l'évaporation et minimiser la dénaturation des protéines.
- Exemples d'utilisation : Conservation des échantillons biologiques, stabilisation des protéines, préparation de milieux de culture.
- Préparation :
1. Ajouter la glycérine à la concentration désirée dans le tampon choisi.
2. Mélanger soigneusement pour assurer une distribution homogène.
- Utilisation :
1. Ajouter comme additif aux tampons pour améliorer la stabilité des échantillons biologiques.
2. Utiliser comme milieu de conservation pour les échantillons biologiques.
5) Tampon de carbonate de sodium :
- Principe : Le carbonate de sodium est utilisé pour maintenir un pH alcalin dans une plage donnée (généralement entre 9 et 11). Il est souvent utilisé pour la dissolution de protéines et la fixation des échantillons biologiques.
- Exemples d'utilisation : Extraction de protéines membranaires, dissolution de protéines dans des solutions tampons, préparation de milieux de culture.
- Préparation :
1. Dissoudre le carbonate de sodium dans de l'eau distillée.
2. Ajuster le pH avec de l'acide chlorhydrique ou de l'hydroxyde de sodium jusqu'à obtenir le pH désiré.
- Utilisation :
1. Utiliser comme tampon alcalin pour la lyse cellulaire ou l'extraction de protéines membranaires.
2. Ajuster le pH pour différentes applications en biochimie et biologie moléculaire.
6) Tampon de Tris-EDTA (TE) :
- Principe : Le tampon de Tris-EDTA est utilisé pour l'extraction, la purification et la conservation de l'ADN. Il contient du Tris pour le maintien du pH et de l'EDTA pour inhiber les enzymes endogènes qui dégradent l'ADN.
- Exemples d'utilisation : Conservation d'échantillons d'ADN, dilution d'échantillons d'ADN pour la PCR, réhydratation de l'ADN lyophilisé.
- Préparation :
1. Dissoudre le Tris et l'EDTA dans de l'eau distillée.
2. Ajuster le pH avec de l'acide chlorhydrique ou de l'hydroxyde de sodium jusqu'à obtenir le pH désiré.
- Utilisation :
1. Utiliser comme tampon pour l'extraction, la purification et la conservation de l'ADN.
2. Diluer les échantillons d'ADN pour la PCR ou d'autres analyses moléculaires.
7) Tampon de carbonate-bicarbonate de sodium :
- Principe : Ce tampon est utilisé pour maintenir un pH alcalin dans une plage donnée, généralement entre 9 et 10. Il est couramment utilisé dans les applications biochimiques et biologiques nécessitant un milieu alcalin.
- Exemples d'utilisation : Réactions d'immobilisation de protéines sur des supports solides, culture de cellules en milieu alcalin, réactions d'hybridation d'acides nucléiques.
- Préparation :
1. Dissoudre le carbonate de sodium et le bicarbonate de sodium dans de l'eau distillée.
2. Ajuster le pH avec de l'acide chlorhydrique ou de l'hydroxyde de sodium jusqu'à obtenir le pH désiré.
- Utilisation :
1. Utiliser comme tampon alcalin dans les réactions biochimiques nécessitant un pH élevé.
2. Préparer des milieux de culture cellulaires avec un pH alcalin pour certaines lignées cellulaires spécifiques.
8) Tampon Tris-glycine :
- Principe : Ce tampon est constitué de Tris et de glycine et est utilisé pour des applications de séparation et d'électrophorèse de protéines. Il offre une bonne résolution et une séparation efficace des protéines dans les gels de polyacrylamide.
- Exemples d'utilisation : Electrophorèse sur gel de polyacrylamide (SDS-PAGE), transfert de protéines sur membrane (Western blot), électrophorèse bidimensionnelle (2D-PAGE).
- Préparation :
1. Dissoudre le Tris et la glycine dans de l'eau distillée.
2. Ajuster le pH avec de l'acide chlorhydrique ou de l'hydroxyde de sodium jusqu'à obtenir le pH désiré.
- Utilisation :
1. Préparer des gels de polyacrylamide pour l'électrophorèse en utilisant ce tampon comme tampon de migration.
2. Utiliser dans les tampons de transfert pour le transfert de protéines sur membrane dans les techniques de Western Blot.
9) SDS (Dodécyl Sulfate de Sodium) :
- Principe : Le SDS est un détergent ionique utilisé dans les applications de biochimie et de biologie moléculaire pour dénaturer les protéines et les rendre linéaires avant l'électrophorèse.
- Exemples d'utilisation : SDS-PAGE (électrophorèse sur gel de polyacrylamide en présence de dodécyl sulfate de sodium), extraction de protéines membranaires.
- Préparation : N/A, le SDS est généralement acheté sous forme de poudre et ajouté directement aux solutions.
- Utilisation :
1. Ajouter à des échantillons protéiques pour les dénaturer avant l'électrophorèse sur gel.
2. Utiliser dans les tampons d'extraction pour solubiliser les protéines membranaires.
10) CTAB (Chlorure de Cetyltriméthylammonium) :
 (source: https://www.slideshare.net/slideshow/different-methods-of-dna-isolation/249634941)
(source: https://www.slideshare.net/slideshow/different-methods-of-dna-isolation/249634941)
- Principe : Le CTAB est un tensioactif cationique utilisé dans l'extraction d'ADN pour lyser les membranes cellulaires et séparer l'ADN des protéines et des polysaccharides.
- Exemples d'utilisation : Extraction d'ADN végétal ou fongique, isolation d'ADN extrachromosomique.
- Préparation : Dissoudre le CTAB dans de l'eau stérile pour obtenir la concentration désirée.
- Utilisation :
1. Utiliser comme composant principal dans les tampons d'extraction pour rompre les membranes cellulaires et libérer l'ADN.
2. Ajouter à des solutions de précipitation pour séparer l'ADN des contaminants protéiques et polysaccharidiques.
11) Triton X100 :
- Principe : Le Triton X100 est un détergent non ionique utilisé pour solubiliser les membranes cellulaires, les lipides et les protéines membranaires.
- Exemples d'utilisation : Lyse cellulaire, extraction de protéines membranaires, préparation de lysats cellulaires pour diverses analyses.
- Préparation : N/A, le Triton X100 est généralement acheté sous forme liquide et utilisé tel quel.
- Utilisation :
1. Ajouter au tampon de lyse pour solubiliser les membranes cellulaires et les composants membranaires.
2. Utiliser comme additif dans les tampons de lavage pour éliminer les contaminants membranaires des échantillons.
12) MgCl2 (Chlorure de Magnésium) :
- Principe : Le MgCl2 est un sel de magnésium couramment utilisé dans de nombreuses réactions enzymatiques comme cofacteur, notamment dans les réactions de polymérisation d'ADN.
- Exemples d'utilisation : Réactions d'amplification d'ADN telles que la PCR, séquençage d'ADN, réactions d'enzyme de restriction.
- Préparation : Dissoudre le MgCl2 dans de l'eau stérile pour obtenir la concentration désirée.
- Utilisation :
1. Ajouter aux mélanges de réaction d'amplification d'ADN pour activer les enzymes telles que les ADN polymérases.
2. Utiliser comme composant dans les tampons de réaction enzymatique nécessitant du magnésium.
13) KCl (Chlorure de Potassium) :
- Principe : Le KCl est un sel de potassium utilisé dans de nombreux tampons et solutions pour ajuster la force ionique, notamment dans les réactions enzymatiques et les réactions d'hybridation d'acides nucléiques.
- Exemples d'utilisation : Extraction d'ADN, réactions d'hybridation d'ARN, électrophorèse sur gel.
- Préparation : Dissoudre le KCl dans de l'eau stérile pour obtenir la concentration désirée.
- Utilisation :
1. Ajuster la force ionique des tampons d'extraction pour favoriser les interactions protéine-ADN.
2. Utiliser comme composant dans les tampons de lavage pour éliminer les contaminants des échantillons.
14) CsCl (Chlorure de Césium) :
(Cette méthode est fastidieuse et difficile à mettre en œuvre car elle nécessite une centrifugation à grande vitesse (100 000 tr/min) pendant plus de 10 heures)
- Utilisation :
1. L'ADN est séparé en fonction de sa densité par centrifugation
2. Lors de la centrifugation à grande vitesse, au point isopycnique où la densité de l'ADN et le gradient (CsCl) deviennent identiques, la bande d'ADN apparaîtra
III) Les techniques biologiques pour lesquelles on utilise les tampons d'extraction
1) Extraction d'acide nucléique de plantes
1) Transformer les plantes lyophilisés ou déshydratés (congelés), en fine poudre.
2) Ajouter la poudre dans une solution tampon d'extraction CTAB (env. 1mL pour 30-50 mg de tissus, le ratio exact dépendant du type de la plante).
3) Incuber la solution obtenu pendant 60 min, à une température entre 55 et 60 °C, en mélangeant occasionnellement.
4) Ajouter une quantité égale de solution de chloroforme et d'alcool isoamyl (24 : 1), et mélanger.
5) Centrifuger, à température ambiante, la solution obtenue, à 1 000-5 000 g, pendant 30 à 50 min.
6) Transférer la phase aqueuse (supérieure), à l'aide d'une pipette large, dans un tube en verre.
7) Ajouter 2.5 volumes de EtOH (-20°C), ou 0.6-1 volume d'isopropanol (-20°C), et mélanger délicatement jusqu'à la précipitation de l'ADN.
- Si les brins d'ADN ne sont pas immédiatement visible, il est possible de laisser la solution reposer quelques jours ou même une nuit entière.
8) Repêcher les brins d'ADN, en utilisant une pipette de Pasteur scellée et incurvée et les transférer dans 10-20 mL, d'une solution de 75% d'EtOH et de 10mM de solution d’acétate d'ammonium. Incuber pendant 20 min, à température ambiante en mélangeant de temps en temps. Répéter une à deux fois.
- Il est possible de faire une pause à cette étape, les brins d'ADN peuvent séjourner même un ou deux jours dans la solution de lavage.
9) Placer l'ADN dans un tube pour microfuge et laisser sécher à l'air libre pendant une quinzaine de minutes.
10) Dissoudre l'ADN dans 200-800 µL de solution stérile de tampon TE (10mM tris-Ha, 1mM EDTA, pH 7.4) et centrifuger dans une microfuge avec une force de 13 000 µg pendant 10 min.
11) Mesurer la concentration en ADN et la réajuster pour avoir une concentration d'environ 0.5-0.7 µg/µL
2) Protéines
3) L'isolation d'ADN plasmidique
Protocole d'Isolation d'ADN Plasmidique par Lyse Alcaline
Matériel requis :
- Eppendorfs stériles
- Centrifugeuse
- Tubes à centrifuger
- Pipettes
- Incubateur à température appropriée
- Réfrigérateur
- Colonne de purification d'ADN ou kit d'extraction d'ADN plasmidique (en option)
Réactifs :
- Solution de Remise en Suspension :
- 50 mM Tris HCl, pH 8
- 10 mM EDTA
- 100 µg/ml RNase A
- Solution de Lyse :
- 0,2 N NaOH
- 1 % de SDS (sodium dodécyl sulfate)
- Acétate de potassium à 3/5 M, pH 6 (solution de neutralisation)
- Isopropanol
- Éthanol à 70 %
- Eau stérile ou solution tampon TE (10 mM Tris, 1 mM EDTA, pH 8)
Procédure :
1. Préparation des Échantillons
- Prélevez 1 à 5 ml de culture bactérienne en croissance logarithmique contenant le plasmide.
- Transférez la culture dans un tube à centrifuger et centrifugez à 3000 x g pendant 5 minutes pour récolter les cellules bactériennes.
2. Lyse des Cellules Bactériennes
- Retirez le surnageant et resuspendez les cellules dans 200 µl de Solution de Remise en Suspension contenant RNase A.
- Incubez les échantillons à 37°C pendant 30 minutes pour digérer l'ARN.
- Ajoutez 200 µl de Solution de Lyse (0,2 N NaOH + 1 % SDS) et mélangez délicatement en inversant plusieurs fois le tube.
- Incubez les échantillons à température ambiante pendant 5 à 10 minutes pour permettre la lyse des cellules.
3. Neutralisation de la Solution de Lyse
- Ajoutez 300 µl d'acétate de potassium (3/5 M, pH 6) à chaque échantillon et mélangez doucement en inversant le tube plusieurs fois.
- Incubez les échantillons sur glace pendant 10 minutes pour permettre la coagulation des protéines et des débris cellulaires.
4. Précipitation de l'ADN Plasmidique
- Centrifugez les échantillons à 12 000 x g pendant 10 minutes à température ambiante pour précipiter l'ADN plasmidique.
- Retirez délicatement le surnageant sans perturber le précipité d'ADN.
5. Lavage de l'ADN Précipité
- Lavez le précipité d'ADN avec 1 ml d'éthanol à 70 % en centrifugeant à 12 000 x g pendant 5 minutes.
- Retirez l'éthanol avec une pipette et laissez sécher le précipité d'ADN à température ambiante pendant quelques minutes.
6. Réhydratation de l'ADN Plasmidique
- Dissolvez l'ADN précipité dans 50 µl d'eau stérile ou de solution tampon TE en incubant à 65°C pendant quelques minutes ou en agitant doucement.
7. Analyse de l'ADN Plasmidique
- Analysez la pureté et la concentration de l'ADN plasmidique isolé en utilisant la spectrophotométrie UV ou d'autres méthodes d'analyse.
4) L'extraction d'ADN à partir de cellules de mammifères
Protocole d'Extraction d'ADN à partir de Cellules de Mammifères
Matériel requis :
- Eppendorfs stériles
- Centrifugeuse
- Tubes à centrifuger
- Pipettes
- Réfrigérateur
Réactifs :
- Solution de Lyse d'ADN :
- 0,1 M EDTA, pH 8,0
- 0,5 % (p/v) de FDS (sodium dodécyl sulfate)
- 10 mM Tris-Cl, pH 8,0
- RNase pancréatique sans DNase à une concentration de 20 μg/ml (à ajouter juste avant utilisation)
Procédure :
1. Préparation des Échantillons
- Récoltez les cellules de mammifères par centrifugation et éliminez le milieu de culture.
- Lavez les cellules avec du PBS (tampon phosphate salin) ou un tampon de votre choix pour éliminer les résidus de milieu de culture.
2. Lyse des Cellules de Mammifères
- Ajoutez 200 µl de Solution de Lyse d'ADN à chaque échantillon de cellules de mammifères.
- Mélangez délicatement en pipetant ou en agitant doucement le tube.
- Incubez les échantillons à température ambiante pendant 5 à 10 minutes pour permettre la lyse des cellules.
3. Traitement avec RNase
- Ajoutez la RNase pancréatique sans DNase à une concentration finale de 20 μg/ml juste avant utilisation.
- Incubez les échantillons à 37°C pendant 30 minutes pour digérer l'ARN.
4. Centrifugation et Collecte de l'ADN
- Centrifugez les échantillons à 12 000 x g pendant 10 minutes pour séparer les débris cellulaires et les protéines de l'ADN.
- Transférez soigneusement le surnageant (contenant l'ADN) dans un nouveau tube propre.
5. Analyse de l'ADN
- Analysez la pureté et la concentration de l'ADN extrait en utilisant la spectrophotométrie UV ou d'autres méthodes d'analyse.
5) L'extraction d'ADN à partir d'échantillons de sang
Protocole d'Extraction d'ADN à partir d'Échantillons de Sang
Matériel requis :
- Eppendorfs stériles
- Centrifugeuse
- Tubes à centrifuger
- Pipettes
- Réfrigérateur
Réactifs :
- Tampon de Lyse pour les Globules Rouges (pH 7,6) :
- 0,155 mol/L NH4Cl
- 10 mmol/L KHCO3
- 0,1 mol/L EDTA (Na2)
- 20 μg/ml de RNase pancréatique sans DNase (ajouter juste avant l'utilisation)
- Tampon d'Extraction à base de CTAB (pH 8,0) :
- 1,5 mol/L Tris, pH 7,6
- 0,4 mol/L Na2 EDTA
- 2,5 mol/L NaCl
- 2 % CTAB (bromure de cétyltriméthylammonium)
- 10 % SDS (dodécylsulfate de sodium)
- β-mercaptoéthanol
- Mélange de chloroforme et d'alcool isoamylique (24:1)
- Isopropanol
- Éthanol à 70 % et 90 %
Procédure :
1. Lyse des Globules Rouges
- Mélangez 1 volume de sang avec 5 volumes de Tampon de Lyse pour les Globules Rouges.
- Incubez à température ambiante pendant 10 minutes pour lyser les globules rouges.
2. Traitement avec RNase
- Ajoutez 20 μg/ml de RNase pancréatique sans DNase au mélange juste avant utilisation.
- Incubez à 37°C pendant 30 minutes pour digérer l'ARN.
3. Lyse Cellulaire avec le Tampon d'Extraction à base de CTAB
- Ajoutez une quantité égale de Tampon d'Extraction à base de CTAB au mélange de sang lyse.
- Incubez à 65°C pendant 10 minutes pour lyser les cellules et dénaturer les protéines.
4. Extraction de l'ADN avec le Mélange de Chloroforme et d'Alcool Isoamylique
- Ajoutez une quantité égale de Mélange de Chloroforme et d'Alcool Isoamylique au mélange et mélangez vigoureusement.
- Centrifugez à 12 000 x g pendant 10 minutes pour séparer les phases.
5. Précipitation de l'ADN avec de l'Isopropanol
- Transférez la phase aqueuse (contenant l'ADN) dans un nouveau tube et ajoutez une quantité égale d'isopropanol.
- Incubez à température ambiante pendant 10 minutes pour précipiter l'ADN.
6. Lavage et Élution de l'ADN
- Lavez l'ADN précipité avec de l'éthanol à 70 % et éliminez l'éthanol résiduel.
- Éluez l'ADN précipité dans de l'eau stérile ou une solution tampon TE.
7. Analyse de l'ADN
- Analysez la pureté et la concentration de l'ADN extrait en utilisant la spectrophotométrie UV ou d'autres méthodes d'analyse.
6) La dissolution des spermatozoïdes et l'extraction d'ADN
Protocole de Dissolution des Spermatozoïdes et d'Extraction d'ADN
Les méthodes d'extraction d'ADN généralement employées pour les cellules somatiques humaines se révèlent inefficaces pour les spermatozoïdes en raison de la compaction nucléaire et de la stabilité de l'ADN propres à ces cellules. Ainsi, un protocole spécifique est nécessaire pour dissoudre les spermatozoïdes et extraire leur ADN de manière appropriée.
Matériel requis :
- Eppendorfs stériles
- Centrifugeuse
- Tubes à centrifuger
- Pipettes
- Réfrigérateur
Réactifs :
- Tampon de Lyse (10x) : (ajouter de l'eau pour obtenir le volume final)
- 5 ml de Tris-HCl à une concentration de 1 M
- 1 ml de NaCl à une concentration de 5 M
- 2,5 ml de MgCl2 à une concentration de 1 M
- 41,5 ml d'eau
- Solution ARN-Plus : 500 µL
- Protéinase K : 50 µL
- Chloroforme : 500 µL
- Éthanol froid pur : 800 µL
- Citrate de sodium 3 M : 40 µL (maintenu à basse température)
- Éthanol à 70 %
Pour la Réhydratation :
- Tris–HCl ou eau distillée et déionisée (ddH2O)
Procédure :
1. Lyse des Spermatozoïdes
- Mélangez 1 volume de Tampon de Lyse (10x) à 9 volumes d'échantillon contenant les spermatozoïdes.
- Incubez à température ambiante pendant 10 minutes pour lyser les spermatozoïdes.
2. Traitement avec Protéinase K
- Ajoutez 500 µL de Solution ARN-Plus et 50 µL de Protéinase K à chaque échantillon.
- Incubez à 37°C pendant 1 heure pour digérer les protéines.
3. Extraction de l'ADN avec du Chloroforme
- Ajoutez 500 µL de chloroforme à chaque échantillon et mélangez vigoureusement.
- Centrifugez à 12 000 x g pendant 10 minutes pour séparer les phases.
- Transférez la phase aqueuse (contenant l'ADN) dans un nouveau tube.
4. Précipitation de l'ADN
- Ajoutez 800 µL d'éthanol froid pur à la phase aqueuse.
- Ajoutez 40 µL de citrate de sodium 3 M à basse température.
- Incubez à -20°C pendant 1 heure ou à température ambiante pendant 10 minutes pour précipiter l'ADN.
5. Lavage et Élution de l'ADN
- Lavez l'ADN précipité avec de l'éthanol à 70 % et éliminez l'éthanol résiduel.
- Éluez l'ADN précipité dans du Tris–HCl ou de l'eau distillée et déionisée.
6. Analyse de l'ADN
- Analysez la pureté et la concentration de l'ADN extrait en utilisant la spectrophotométrie UV ou d'autres méthodes d'analyse.
Conclusion
En conclusion, les tampons d'extraction jouent un rôle crucial dans la purification et la préservation de l'ADN et des protéines lors des procédures d'analyse biologique. Le choix approprié du tampon et le respect des protocoles spécifiques garantissent le maintien du pH optimal, la lyse efficace des cellules, la séparation des composants cellulaires et des contaminants, ainsi que la préservation de l'intégrité des molécules ciblées. Ces tampons offrent une variété de compositions et de pH adaptés à différentes applications, ce qui en fait des outils indispensables pour les techniques d'extraction et d'analyse en biologie moléculaire et cellulaire.
Les liens qui peuvent être intéressants ... :
DNA extraction methods using magnetic beads
DNA extraction using anion exchange resins